As the coronavirus pandemic has swept across the U.S., in addition to tracking the number of COVID-19 daily cases, there is a worldwide scientific community engaged in tracking the SARS-CoV-2 virus itself.

Efrem Lim leads a team at Arizona State University that looks at how the virus may be spreading, mutating and adapting over time.
To trace the trail of the virus worldwide, Lim’s team is using a new technology at ASU’s Genomics Facility called next-generation sequencing to rapidly read through all 30,000 chemical letters of the SARS-CoV-2 genetic code, called a genome.
Each sequence is deposited into a worldwide gene bank, run by a nonprofit scientific organization called GISAID. To date, over 16,000 SARS-CoV-2 sequences have been deposited in GISAID’s EpiCoVTM Database. The sequence data shows that SARS-CoV-2 originated from a single source from Wuhan, China, while many of the first Arizona cases analyzed showed travel from Europe as the most likely source.
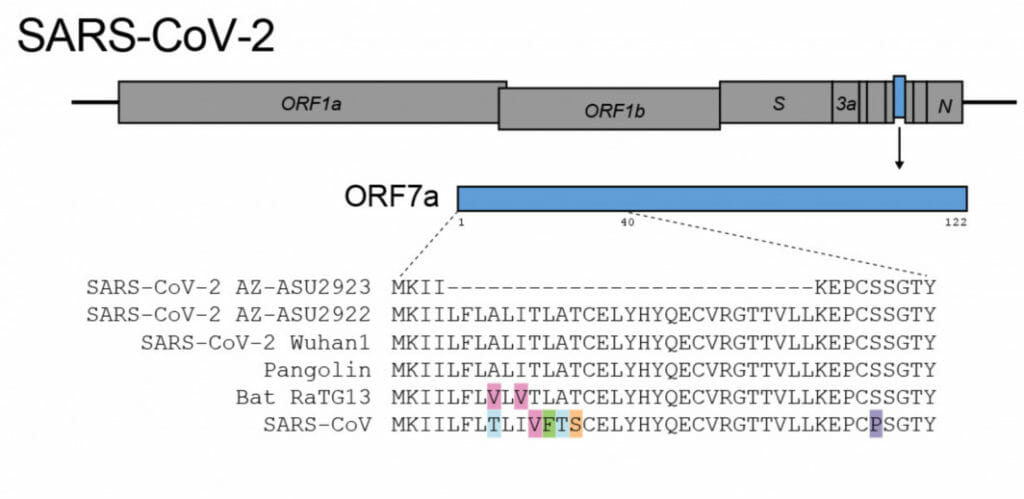
Now, using a pool of 382 nasal swab samples obtained from possible COVID-19 cases in Arizona, Lim’s team has identified a SARS-CoV-2 mutation that had never been found before — where 81 of the letters have vanished, permanently deleted from the genome.
The study was published in the online version of the Journal of Virology.
Lim says as soon as he made the manuscript data available on a preprint server medRxiv, it attracted worldwide interest from the scientific community, including the World Health Organization.
“One of the reasons why this mutation is of interest is because it mirrors a large deletion that arose in the 2003 SARS outbreak,” said Lim, an assistant professor at ASU’s Biodesign Institute.
During the middle and late phases of the SARS epidemic, SARS-CoV accumulated mutations that attenuated the virus. Scientists believe that a weakened virus that causes less severe disease may have a selective advantage if it is able to spread efficiently through populations by people who are infected unknowingly.
Teasing apart what exactly this means is of profound interest to Lim and his colleagues. The ASU research team includes LaRinda A. Holland, Emily A. Kaelin, Rabia Maqsood, Bereket Estifanos, Lily I. Wu, Arvind Varsani, Rolf U. Halden, Brenda G. Hogue and Matthew Scotch.
The ASU virology team had been set up to perform research on seasonal flu viruses, but when the third case of COVID-19 was found in an Arizona individual on Jan. 26, 2020, they knew they had all technical and scientific prowess to rapidly pivot to examining the spread of SARS-CoV-2.
“This was the scientific opportunity of a lifetime for ASU to be able to contribute to understand how this virus is spreading in our community,” Lim said. “As a team, we knew we could make a significant difference.”
All the positive cases show that the SARS-CoV-2 viral genomes were different from each other, meaning they were independent from each other. This indicates that the new cases were not linked to the first Arizona case in January, but were the result of recent travel from different locations.
In the case of the 81-base pair mutation, because it has never been found before in the GISAID database, it could also provide a clue into how the virus makes people sick. It could also form a new starting point for other scientists to develop antiviral drugs or formulate new vaccines.
SARS-CoV-2 makes accessory proteins that help it infect its human host, replicate and eventually spread from person to person. The genome deletion removes 27 protein building blocks, called amino acids, from the SARS-CoV-2 accessory protein ORF7a. The protein is very similar to the 2003 SARS-CoV immune antagonist ORF7a/X4.
The ASU team is now hard at work performing further experiments to understand the functional consequences of the viral mutation. The viral protein is thought to help SARS-CoV-2 evade human defenses, eventually killing the cell. This frees up the virus to infect other cells in a cascading chain reaction that can quickly cause the virus to make copies of itself throughout the body, eventually causing serious COVID-19 symptoms eight to 14 days after the initial infection.
Lim points out that only 16,000 SARS-CoV-2 genomes have been sequenced to date, which is less than 0.5% of the strains circulating. There are currently more than 3.5 million confirmed COVID-19 cases worldwide.
Lim’s group has teamed up with TGen, UArizona and Northern Arizona University to continue tracking different genetic strains of the new coronavirus. Together, the newly formed Arizona COVID-19 Genomics Union (ACGU) hopes to use big data analysis and genetic mapping to give Arizona health care providers and public policy makers an edge in fighting the growing pandemic.
The work was supported by NSF STC Award 1231306, NIH grants R01 LM013129, R00 DK107923, the J.M. Kaplan Foundation’s One Water One Health, Arizona State University Foundation project 30009070), and ASU Core Facilities Seed Funding.